Como un primer ejemplo de técnicas de perturbación aplicadas a modelos de átomos hidrogenoides, supóngase que se tiene un núcleo con carga Ze y un solo electrón. Hay una solución analítica exacta para lo primero que será estudiado a continuación, pero de cualquier modo el cálculo perturbativo nos permitirá comparar qué tanto se aproxima la aproximación perturbativa a la solución exacta. La energía del estado base del electrón para un átomo de hidrógeno en el modelo de Bohr en el sistema de unidades MKS-SI es:

mientras que la energía del estado base para un átomo hidrogenoide con número atómico Z y por ende con carga nuclear Ze es:

Supóngase ahora que el núcleo sufre un decaimiento β emitiendo un electrón, y como consecuencia de ello la carga nuclear positiva aumenta a:
(Z + 1) e
La energía del estado base será ahora:

A continuación, calcularemos la nueva energía que resulta como consecuencia de lo que consideraremos una perturbación, obtenida mediante la teoría de las perturbaciones. El Hamiltoniano H0 del estado original (no-perturbado) es:

siendo la función de onda radial para el estado base:

en donde la condición de normalización para la función de onda radial debe cumplir con la relación:

El nuevo Hamiltoniano H incorporando los efectos de la perturbación a causa del decaimiento beta es:

La perturbación es solamente 1/Z veces el potencial original en todo punto, por lo que en una corrección perturbativa de primer orden se puede tomar a λ como 1/Z. La aproximación perturbativa de primer orden nos proporciona la nueva energía E como:

en donde E0 es la eigenenergía no-perturbada, U es la perturbación, y se supone de antemano que la función de onda ψ0 está normalizada. Se tiene entonces lo siguiente:

en donde para la evaluación de la integral se ha usado la siguiente forma tomada directamente de las tablas de integrales:

Por lo tanto:

De acuerdo al cálculo que se acaba de llevar a cabo usando la teoría de las perturbaciones, la corrección de primer orden que se ha calculado es 2/Z veces la energía original, o sea 2λE0. La respuesta analítica correcta (exacta) para la energía E es:

Comparando la diferencia entre la solución exacta con la obtenida empleando la teoría de las perturbaciones, podemos ver que tal diferencia es:

Para el cálculo de las energías de los estados cuánticos de átomos con un solo electrón en la capa exterior sometido a la acción de un campo de carga Ze, en los cursos introductorios se supone por razones de simplicidad que el núcleo atómico es una carga puntual. Sin embargo, ésto es ilusorio, ya que geométricamente hablando un punto carece de dimensiones y en el mundo físico tal idealización vendría siendo una referencia a algo que no puede tener existencia en el mundo real. Debemos aceptar el hecho de que los núcleos reales tienen dimensiones finitas, y por lo tanto el potencial fuera del núcleo es -Ze/4πε0 solamente hasta la superficie del núcleo (yendo desde la lejanía hasta la capa nuclear), y se desvía de ésta cantidad en su interior. Calcularemos el efecto del tamaño finito del núcleo mediante la teoría de las perturbaciones.
Por principio de cuentas, supondremos que el núcleo es una esfera con carga uniforme de radio R. Empleando el teorema de Gauss en la electrostática, y suponiendo una densidad uniforme ρ de carga eléctrica en el interior del núcleo, no es difícil demostrar que en el interior del núcleo el campo eléctrico debe aumentar linealmente desde cero en el origen hasta Ze/4πεR2 en la superficie. Por lo tanto, el campo eléctrico ℰ en el interior del núcleo atómico está dado por la relación:

como lo ilustra la siguiente figura:

Tenemos por otro lado la ecuación que nos define el campo eléctrico ℰ en función del potencial eléctrico Φ (precaución: jamás se confunda por ningún motivo el potencial eléctrico Φ usualmente expresado en volts con la función de la energía potencial V(r) en la ecuación de Schrödinger usualmente expresada en joules o ergs):

Usando dicha ecuación definitoria se tiene entonces lo siguiente:

La energía potencial U(r) medida en joules, en función del potencial eléctrico Φ expresado en volts, es:

Para un electrón con su carga eléctrica negativa la energía potencial en el interior del núcleo será entonces:

La energía potencial del electrón en el caso de una carga puntual es:

y por lo tanto la perturbación está dada en todo el espacio físico disponible por:

La corrección de primer orden de acuerdo a la teoría de las perturbaciones es:

y por lo tanto la corrección de primer orden para la energía del estado base debida al tamaño finito del núcleo se obtiene del modo siguiente:

Para poder llevar a cabo la integración, necesitamos alguna aproximación al radio R del núcleo. El radio del núcleo se obtiene con bastante buena aproximación mediante la fórmula:

en donde r0.=.1.3×10-13.cm. y A es el número de la masa atómica. Aún para los núcleos más pesados, por ejemplo el plomo para el cual A.=.208 y Z.=.82, R no es mayor que 10-12.cm., y el parámetro del radio a0/Z siempre es mayor que 10-12.cm. Por lo tanto, el término exponencial en la integral puede igualarse a la unidad:

y así la integración se puede llevar a cabo de la manera siguiente:

Empleando nuevamente la fórmula empírica dada arriba que nos proporciona una aproximación razonable para el radio R del núcleo para los elementos diversos, se obtiene el siguiente resultado para el radio del núcleo de un elemento ligero, el neón (Z.=.10, A.=.20):
R = (1.3×10-13 cm.) · (20)1/3
R = 3.5287429×10-13 cm.
Habiendo obtenido el radio R del núcleo del neón, usamos la expresión derivada arriba para calcular la corrección de primer orden E(1) para la energía del estado base del neón que resulta ser:
E(1) = 4.9×10-4 eV
Resulta conveniente poner ésta corrección energética de primer orden E(1) en función de la energía del estado base del neón. Podemos calcular la energía del estado base del neón recurriendo primero a la conocida fórmula para el átomo de Bohr que nos proporciona los niveles energéticos correspondientes a cada número cuántico n para un átomo hidrogenoide cuyo número atómico es Z:

De acuerdo a ésta fórmula, la energía del estado base para un elemento hidrogenoide de número atómico Z que se encuentra en el estado fundamental n.=.1 será:

en donde se ha usado la energía del estado base del hidrógeno que ya se sabe que es 13.6.eV. En conformidad con ésta relación, la energía del estado base para el neón será (prescindiremos del signo negativo ya que no lo necesitamos para el propósito presente):
Ebase = (10)2 · (13.6 eV)
Ebase = 13.6×102 eV
Es fácil verificar entonces que la corrección perturbativa de primer orden para el neón bajo el modelo que está siendo utilizado, en función de la energía del estado base del neón, será:
E(1) = 4.9×10-4 eV
E(1) = (3.6×10-7) (13.6×102) eV
E(1) = (3.6×10-7) Ebase
R = (1.3×10-13 cm.) · (208)1/3
R = 7.7024898×10-13 cm.
Habiendo obtenido el radio R del núcleo del plomo, usamos la expresión derivada arriba para calcular la corrección de primer orden E(1) para la energía del estado base del plomo que resulta ser:
E(1) = 10.4 eV
Del mismo modo, la energía del estado base para el plomo será:
Ebase = (82)2 · (13.6 eV)
Ebase = 91,446 eV
Es fácil verificar entonces que la corrección perturbativa de primer orden para el plomo bajo el modelo que está siendo utilizado, en función de la energía del estado base del plomo, será:
E(1) = 10.4 eV
E(1) = (1.14×10-4) (91,446) eV
E(1) = (1.14×10-4) Ebase
Estamos ya en condiciones de poder efectuar comparaciones entre las diferencias que resultan de la aplicación del modelo especificado a un elemento ligero y a un elemento pesado. Comparando los resultados obtenidos para ambos elementos, se concluye que para elementos ligeros el efecto es totalmente despreciable, mientras que para elementos pesados el efecto, aunque pequeño, es susceptible de poder ser detectado. El caso es totalmente distinto si se considera un sistema formado cuando el átomo captura un mesón μ con carga negativa. Tal átomo muónico tiene un sistema de niveles de energía similar en todos sentidos al de un átomo con un solo electrón, con excepción del parámetro del radio a0(μ) que se reduce en comparación con el de un electrón, por la razón de sus masas. El mesón μ tiene una masa equivalente a 207 veces la del electrón, y por lo tanto:

Cuando Z es mayor que aproximadamente 50 la función de onda del estado base del núcleo puntual tiene un parámetro de radio menor que el radio nuclear. La corrección para el neón es de aproximadamente 1.5%.
Ahora consideraremos un problema en el cual se supone que un electrón se mueve en el potencial:

Considerando la desviación con respecto a un potencial de Coulomb como una perturbación, el objetivo es calcular la energía del electrón en el estado base y tratar de responder la siguiente pregunta: ¿en qué intervalo de valores de r0 se esperaría obtener una respuesta confiable?
Si como perturbación se va a tomar la desviación con respecto al potencial de Coulomb, la perturbación será entonces:

El cambio en la energía del estado basal a causa de lo que estamos considerando como una perturbación en una corrección a un primer orden será:

No aparece mención alguna al número atómico Z en las fórmulas del modelo, por lo que se sobreentiende que el modelo corresponde al de un átomo de hidrógeno para el cual Z.=.1, con lo cual la función de onda radial:

se vuelve simplemente:

De éste modo, se tiene el siguiente cálculo:

Llevando a cabo la integración parcial se obtiene:

De lo que se ha obtenido se concluye que para que la corrección perturbativa de primer orden E(0) sea menor de un 10 por ciento de la energía del estado base, por ejemplo, el término 2r0/a0 deberá ser mayor que cinco.
Como un cuarto ejemplo de modelos perturbativos aplicados a átomos hidrogenoides, supóngase que el protón en un átomo de hidrógeno es una capa esférica delgada con radio r0, creando una energía potencial de (obsérvese que se trata de un potencial uniforme y constante en toda la región interior):

para el interior de la capa esférica, y de (obsérvese que se trata de un potencial variable en toda la región exterior que va cayendo en razón inversa al radio)::

para el exterior de la capa esférica. Emplearemos la teoría de las perturbaciones para estimar el cambio de la energía del estado base del átomo de hidrógeno que ocasionará la especificación de éste modelo.
La perturbación, como está dada, puede simbolizarse del siguiente modo:
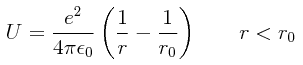
siendo U.=.0 en todos los demás casos (o sea en el exterior de la casco esférico delgado). Procediendo en forma parecida a los casos tratados arriba, y haciendo a un lado el factor e2/4πε0 para concentrarnos exclusivamente en el proceso de integración, vemos que se requiere evaluar:

Para poder llevar a cabo la integración entre los límites indicados, se emplea el siguiente resultado:

Efectuando la integración y agrupando términos, se obtiene:

Se puede especificar, en principio, una cantidad prácticamente infinita de modelos teóricos diversos para los átomos hidrogenoides, algunos mucho más complejos que otros, con la idea en mente de echar mano posteriormente de la teoría de las perturbaciones para obtener soluciones aproximadas. Lo difícil no es inventar modelos teóricos según se nos vengan a la imaginación. Lo difícil es justificar de alguna manera los términos y complejidades que terminemos metiendo en cada modelo que se nos ocurra. Es algo parecido a lo que sucedió con la ecuación general de los gases ideales, la cual fue refinada posteriormente con la ecuación de Van der Waals en la cual el físico holandés Johannes van der Waals introdujo dos términos correctivos tanto en la presión como el volumen del gas para producir una ecuación que podía resumir mejor el comportamiento de los gases reales y la desviación de éstos con respecto al modelo del gas ideal. Van der Waals no solo introdujo tales términos correctivos, sino que dió una justificación teórica a ambas correcciones, las cuales inclusive pueden ser justificadas en tratados superiores de termodinámica, logrando con ello que la ecuación de van der Waals no sea simplemente una fórmula empírica como tantas otras.

en donde δl es una constante para un valor determinado de l e independiente de n. A δl se le conoce como el defecto cuántico (no confundir con el mismo término empleado en la ciencia de los rayos láser, aquí lo estamos aplicando al caso de los átomos que se ajustan a las predicciones de las rayas espectrales predichas por la fórmula de Rydberg). El defecto cuántico es una cantidad puramente empírica sin una derivación teórica formal, aunque permite la penetración de las capas interiores del átomo por el electrón de valencia. Así, mientras que en el sodio se tiene para los niveles de onda s:

para los niveles de onda p del mismo sodio se tiene:

La siguiente tabla proporciona los defectos cuánticos para el espectro del átomo de sodio:
| Término | n = 3 | n = 4 | n = 5 | n = 6 | n = 7 | n = 8 | |
| l = 0 |
s
|
1.373 | 1.357 | 1.352 | 1.349 | 1.348 | 1.351 |
l = 1
|
p
|
0.883 | 0.867 | 0.862 | 0.859 | 0.858 | 0.857 |
| l = 2 |
d
|
0.010 | 0.011 | 0.013 | 0.011 | 0.009 | 0.013 |
| l = 3 |
f
|
-
|
0.000 | -0.001 | -0.008 | -0.012 | -0.015 |
Para demostrar cómo difieren las energías dadas por la ecuación mostrada arriba dentro de la cual se incorpora el defecto cuántico, se desarrolla la ecuación en potencias ascendientes de δl:

El primer término es el valor no-perturbado para el hidrógeno en el modelo atómico planetario de Bohr, mientras que el resto es la corrección que se debe aplicar. Para valores grandes de n el segundo término, proporcional a 1/n3, es evidentemente el término dominante de la corrección. Para pequeños valores de n y l, por ejemplo n.=.3, l.=.0 en el sodio, el segundo término, aunque mayor que los términos más elevados, no es mayor que el tercer término. En estos casos, sin embargo, la corrección es tan grande, que no podemos esperar que la teoría de las perturbaciones de primer orden, comenzando con las funciones de onda del átomo de hidrógeno, dé más de una indicación cualitativa de la corrección. Así se ha logrado entender cuando menos parcialmente por qué la ecuación de arriba es una buena representación de las energías de los niveles cuando se puede demostrar que la teoría de las perturbaciones nos dá una corrección aproximadamente proporcional a 1/n3.
Con la incorporación del defecto cuántico, tenemos una fórmula de Rydberg modificada que se puede emplear en átomos polielectrónicos como el sodio para predecir las líneas espectrales que se esperan:

Para el sodio, por ejemplo, se tiene usando valores como los de la tabla dada arriba:

lo cual concuerda bastante bien con la ubicación de las líneas-D del espectro del sodio que está dominado por el brillante doblete situado en las longitudes de onda de a 588,9950 y 589,5924 nanómetros.
El principal propósito de la Teoría de las Perturbaciones fue abrir la puerta al entendimiento del comportamiento de todos los demás elementos diferentes al hidrógeno desde la perspectiva de la Mecánica Cuántica. El único elemento de toda la Tabla Periódica cuyo operador Hamiltoniano H de energía que tiene una solución exacta para la ecuación diferencial de Schrödinger es el más sencillo de todos, el hidrógeno. El siguiente elemento más complicado, el helio, con dos electrones en órbita interactuando entre sí, es soluble pero con un aumento considerable en la complejidad matemática. Y para el resto podemos dejar atrás cualquier esperanza de poder obtener soluciones exactas, necesariamente se tiene que recurrir a aproximaciones, manejadas como perturbaciones, para poder extraer deducciones entendibles que concuerden con las observaciones de las propiedades físicas, y muy en especial las propiedades químicas, de los elementos. Entendiblemente, una vez resuelto en forma exacta el problema mecánico-cuántico del átomo de hidrógeno, el siguiente paso consiste en extender los resultados obtenidos hacia los átomos más parecidos al hidrógeno, los átomos hidrogenoides.
La configuración del estado base de los metales alcalinos (Li, Na, K, Rb, Cs) consiste de un solo electrón de valencia en un estado s, fuera de una capa cerrada, como puede verse en la siguiente tabla que nos muestra las configuraciones electrónicas de los átomos:
Z
|
Elemento | 1s | 2s | 2p | 3s | 3p | 3d | 4s | 4p | 4d | 4 f | 5s | 5p | 5d | 6s |
| 1 |
H
|
1
|
|||||||||||||
| 3 |
Li
|
2
|
1
|
||||||||||||
| 11 |
Na
|
2
|
2
|
6
|
1
|
||||||||||
| 19 |
K
|
2
|
2
|
6
|
2
|
6
|
1
|
||||||||
| 37 |
Rb
|
2
|
2
|
6
|
2
|
6
|
10
|
2
|
6
|
1
|
|||||
| 55 |
Cs
|
2
|
2
|
6
|
2
|
6
|
10
|
2
|
6
|
10
|
2
|
6
|
1
|
Es común ver en textos de química una figura como la siguiente para ilustrar el concepto idealizado del electrón de valencia solitario afuera de un núcleo que está recubierto por capas cerradas:

en donde cada una de las flechas apunta hacia el electrón de valencia solitario, aunque esta figura es una sobresimplificación que ignora la existencia de subcapas como p, d y f predichas por la Mecánica Cuántica.
Desde el punto de vista de la química, en el nivel más elemental la valencia se define como el número de átomos de hidrógeno que son desplazados por, o se combinan con, un átomo del elemento que se está considerando. Como el hidrógeno tiene sólo un electrón, es de esperarse que los átomos que tengan un electrón fuera de una capa cerrada ligada fuertemente sean monovalentes. Así sucede en realidad, son los metales alcalinos. De igual forma es de esperarse que los átomos que necesiten un electrón para completar una capa sean monovalentes. Estos elementos, los halógenos, tienen potenciales de ionización elevados y por lo tanto no comparten con facilidad sus electrones con otros átomos, pero pueden recibir un electrón adicional para completar la capa, y no más de uno, porque un segundo electrón iría a una órbita más elevada. De la misma manera los átomos con dos electrones fuera de una capa cerrada o que requieren dos electrones más para completar una capa cerrada suelen ser divalentes. Más allá de este punto, la variedad de modos en que pueden ordenarse los electrones significa generalmente que puede existir más de una valencia. Por ejemplo, hay tres óxidos de nitrógeno distintos, y dos de carbono, y la valencia puede depender de la naturaleza de otros átomos con que el elemento se combine. No obstante, se encuentra, como es de esperarse, que los elementos con el mismo número de electrones externos y con estructuras similares de capas internas, tengan propiedades químicas muy similares.
Los estados excitados que vamos a estudiar son aquellos en los que el electrón de valencia se promueve a estados más elevados. Para que los argumentos empleados sean específicos se estudiará en particular uno de los metales alcalinos, el sodio, pero estos argumentos pueden aplicarse asimismo a todos los metales alcalinos con los cambios correspondientes en los valores numéricos y números cuánticos.
El sodio Na tiene once electrones. Diez de ellos llenan las capas n.=.1 y n.=.2 y el electrón de valencia es estado base es un electrón 3s. Los estados excitados que consideraremos son aquellos en que las capas n.=.1 y n.=.2 permanecen llenas y el electrón de valencia se promueve a los estados 3p y 3d, y a los estados 4s, 4d y 4f, y así sucesivamente.
Como primera aproximación se supondrá que la función de onda del electrón de valencia en cualquiera de los estados anteriores (incluyendo al estado base) es la misma que para un electrón con los mismos números cuánticos en el átomo de hidrógeno. Es necesario primero justificar esta suposición. La suposición se justificará si se demuestra que en la región ocupada por la función de onda del electrón de valencia, el potencial que dicho electrón experimenta es solamente el potencial de Coulomb. En la siguiente figura se grafica el cuadrado de la función de onda radial |u(r)|2 para los estados 3s y 3p del hidrógeno:

Se ve que el grueso de la probabilidad se encuentra más allá de r.=.4a0 en donde a0 es el radio de Bohr 4πε0ħ2/(me2) .. Como los estados más elevados están más debilmente ligados, se extenderán hacia afuera.
A grandes distancias, el potencial que experimenta el electrón de valencia será el potencial de Coulomb debido a una sola carga, porque los diez electrones internos con carga eléctrica negativa compensarán diez de las once cargas positivas del núcleo. Deseamos estimar a qué distancia se hace incompleto el anterior apantallamiento. Puede hacerse de la siguiente manera. Se ha señalado en entradas previas que a grandes distancias todas las funciones de onda de los electrones disminuyen según e-αr en donde α.=.(2mE/ħ2)1/2 y E es la energía de enlace del electrón. La energía de los electrones en la capa interna será del orden del segundo potencial de ionización que es 47 voltios para el sodio. Así, tenemos que E.≈.47 eV y por lo tanto α es aproximadamente 2/a0. La densidad de carga debida a los electrones internos disminuye según el siguiente factor:

porque es proporcional al cuadrado de la función de onda. Como hay diez electrones internos, es necesario que el factor sea bastante pequeño, por ejemplo del orden de 10-3, antes de poder decir con certeza que la cantidad de carga que se encuentre a mayores distancias es despreciable y el efecto de apantallamiento de los electrones internos es completo. Podemos apreciar que esta condición se logra cuando r.=.2a0 (e-8.=.0.3×10-3). Por lo tanto, el grueso de la función de onda del electrón de valencia se encuentra realmente en una región en que el potencial que experimenta es el debido a una sola carga.
Por consiguiente, para encontrar el espectro de niveles de energía del sodio se comienza con funciones de onda y energías dadas por el átomo de hidrógeno, y empleamos la teoría de las perturbaciones para calcular la variación de la energía debida a la desviación del potencial a partir de la forma Coulombiana para pequeñas distancias. Si se inspecciona la porción inicial de las funciones de onda 3s, 3p y 3d en una gráfica un poco más completa como la que se ha puesto que muestra las funciones de onda 3s y 3p, junto con el potencial de perturbación U debido a la decreciente eficacia del apantallamiento de la carga nuclear por los electrones de la coraza, las características cualitativas de lo que sucede se vuelven evidentes. La onda d con l.=.2 es muy pequeña en toda la región en que la perturbación es significativa, y por consecuencia la energía de este nivel casi no cambia con respecto al valor para el hidrógeno. La onda p con l.=.1 tiene algo más de su función de onda dentro del potencial de la perturbación, debido al máximo que se produce en r.=.a0 (véase la figura de arriba), en tanto que la solución de la onda s tiene todo el primer máximo dentro del potencial de la perturbación.
Por lo tanto, el efecto de la desviación con respecto al potencial Coulombiano es dividir la degeneración de los niveles n.=.3 con distintos momentos angulares orbitales. O, empleando el lenguaje propio de la Teoría de las Perturbaciones, se rompe la degeneración de los estados traslapados produciéndose una separación energética entre lo que eran los estados degenerados. No es sorprendente que el estado con momento angular más elevado seea muy poco afectado, porque la fuerza centrífuga debida a su momento angular lo aleja de la región en que se produce la perturbación. Lo anterior se puede apreciar en una figura en la cual se grafique el potencial radial efectivo total:

para el potencial Coulombiano y el potencial del sodio, en ambos casos con l.=.2. Se encuentra que los dos casos no se desvían significativamente uno del otro, hasta que estamos bien adentrados en la región clásicamente prohibida.
Los estados de momento angular bajo tienen elevada probabilidad de que el electrón esté muy cerca del núcleo, en donde se verá fuertemente afectado por la carga nuclear, y consiguientemente su nivel energético disminuirá.
Estas características cualitativas se aplican con la misma fuerza a los niveles más elevados del sodio. En virtud de que la función de onda radial u(r) se comporta de la siguiente forma cuando la distancia radial al origen r es pequeña:

y tomando en cuenta la forma en que la función de onda se inicia muy cerca del origen, la función de onda radial está por lo tanto determinada por el valor del momento angular, por lo cual todos los niveles de onda s se desplazan substancialmente, todos los niveles p se desplazan moderadamente, y los niveles con l.=.2 casi no son afectados. Esto se puede apreciar mejor en la siguiente figura en donde se muestran los niveles de energía del sodio, junto con las posiciones de los niveles correspondientes del hidrógeno. La situación de los demás metales alcalinos es parecida, con excepción del rubidio Rb y el cesio Cs, en los que se observan grandes corrimientos en los niveles de onda d. Los niveles f permanecen esencialmente sin desplazarse en toda la serie de los metales alcalinos. Los niveles de energía del átomo de sodio se muestran en electrón-voltios para l.=.0,1,2, mientras que a la derecha se muestran los niveles del átomo de hidrógeno para n de 2 a 6:

